الثلاسيميا
الثلاسيميا :
مرض الدم الوراثي غير المعدي الإنحلالي و من أكثر أمراض الوراثة إنتشارا ًفي العالم ولا يفرق في الإصابة بين جنس وأخر ولا بين ذكر أو أنثى . أول مكتشف له هو الطبيب كولي وذلك عام 1925 . ويطلق على المرض أيضا ً تسمية ( أنيميا حوض البحر الأبيض المتوسط ) وكلمة أنيميا اليونانية الأصل تتكون لغويا ً من ـ نيميا ـ والتي تعني الدم وحرف الA بمعنى النفي لتصبح الترجمة الحرفية ـ لا دم ـ أي فقر الدم . كما أسلفت فإن المرض ينتشر في جميع أنحاء العالم , ولكن إنتشاره بنسبة أكبر في بعض البلدان مثل بلدان حوض البحر الأبيض المتوسط ومن هنا جاءت التسمية المرادفة له . وتجدر الإشارة أن الإحصائيات الأخيرة اوضحت أن نسبة الإصابة بالمرض في الخليج العربي بلغت ال 5% من مجموع السكان .
وهذا المرض يصيب بالتحديد تكوين الدم وكرياتة الحمراء ويؤدي بشكل رئيسي إلى إنخفاض نسبة الهموغلوبين . ولمزيدا ً من الإيضاح نلقي نظرة علمية سريعة لنرى أن لدى الجنين في بطن امه هيموغلوبين جنيني HBF والذي يتحول في فترات لاحقة إلى هيموغلوبين البالغين HBA ويسمى أيضا ً( خضاب الدم ) وتتكون كلمة الهيموغلوبين الإنكليزية من ـ هيم ـ أي الحديد والصبغة وغلوبين أي البروتين .
وبالرجوع السريع إلى علم الوراثة نرى ان الأصحاء من البشر يملكون جينين سليمين يتحكمان في صنع ونقل الهيموغلوبين , أما الأشخاص حاملي مرض الثلاسيميا فيملكون جينا ً واحدا ًطبيعيا ً والثاني يحمل الصفة المريضة .في الوقت الذي يحمل مرضى الثلاسيميا جينان مريضان موروثان من كلا الأبوين .
ولأكمال الأيضاح والتقريب العلمي أذكٍر قارئئ اللبيب بأن خلايا الدم الحمراء تتكون وتتولد غالبا ًفي نخاع العظام وتعيش 120 يوما ثم تموت لتولد كريات جديدة أخرى ومهمتها الرئيسية نقل وتوزيع الأوكسجين إلى مختلف أعضاء وأنسجة الجسم وذلك بواسطة ربطه على الهيموغلوبين الموجود في تلك الكرية الحمراء .
التصنيف والأعراض والمضاعفات
إختلفت التصانيف ولكن للتبسيط وسهولة الفهم من الممكن مجازا ً تصنيف المرض إلى الحالات والأصناف التالية فالصغرى منها عادة ما يكون الشخص حاملا ً للمرض غير مصابا ًبه ولا توجد لديه أية أعراض أو شكوى مرضية , والمتوسطة وفيها يكون الفرد مصابا ًبالمرض ً, لكن بأعراض خفيفة مثل فقر الدم والشحوب والضعف العام البسيط , وفي الحالتين أو الصنفين المذكورين سابقا ً يكون الفرد قادرا ًعلى نقل المرض لأبنائه . أما في حالة الإصابة بالثلاسيميا الكبرى فتكون الأعراض شديدة الوطئة ومنها أذكر لا حصرا ً تاخر الأطفال المصابون بالمرض في مراحل النمو, فقر الدم الشديد وإنخفاض نسبة الهيموغلوبين نتيجة لتكسر كريات الدم الحمراء المستمر ومايتبع ذلك من شحوب الوجه وإصفراره وفقدان الشهية والنحول العام , إضطراب النوم وضعف المناعة ومقاومة الأمراض المعدية والإنتقالية . كما يتسبب تكسر كريات الدم الحمراء بزيادة نسبة الحديد في الجسم وترسبه في مختلف الأنسجة ويؤدي ذاك التكسر إلى تضخم الكبد والطحال,أما تضخم نخاع العظام فيتبعه بروز عظام الوجه وبشكل ملحوظ .وغالبا ًما يصاحب المرض فشل غدة البنكرياس مما يؤدي للاصابة بداء السكري ويصاحبه أيضا ًقصور في الوظيفة الجنسية والغدة الدرقية . وتعد الإنعكاسات النفسية على الطفل المصاب والمتاعب الحياتية و الإقتصاديةعلى عائلته من أهم المضاعفات المرافقة للمرض والناتجة عنه .
الوقاية من خلال التأكيد على ضرورة إجراء الفحص الطبي للراغبين بالزواج للتأكد من خلو الطرفين من حمل صفة المرض وهنا تجدر الإشارة إن سلامة أحد الزوجين تكفي لأنجاب أطفال أصحاء خالين من التلاسيميا والنقطة المهمة الثانية للوقاية من الإصابة هي تجنب زواج الأقارب من بعضهم البعض.
التشخيص
سهل ومتوفر ويتم بإجراء فحص الدم المختبري والمعروف بالترحيل الكهربائي للهيموغلوبينُ Electrophoresis Hemoglobin
العلاج
يشمل نواحي ومفاصل متعددة ومتنوعة أوردها بإختصار بالنقاط التالية :
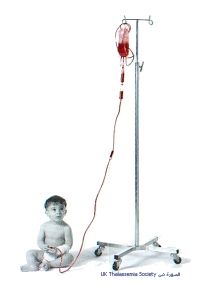
•1. نقل الدم المتكرر والتعويضي للحفاظ على مستوى مقبول للهيموغلوبين , لكن تكرار نقل الدم يسبب زيادة تحرر وترسب الحديد لدرجة الضرر ببعض أجهزة الجسم وتعطيلها مما يتطلب تناول المريض لمادة الدسفيرال للتخلص من ذلك الحديد الزائد وكذلك إعطاء المريض مادة الفوليك اسد للمساعدة بإنتاج كريات الدم الحمراء
•2. تناول فيتامين D وتجنب المواد الغذائية الغنية بالحديد كالكبد والسبانغ
•3. المعالجة الجراحية وتتضمن إستئصال الطحا ل وزراعة نخاع العظم .
وقد تمت اول زراعة ناجحة لنخاع عظم طفل عمره سنة ونصف في عام 1981 بعد التطابق التام للأنسجة للمريض والمتبرع .
ختام المسك تقبلوا صادق دعائي بدوام الصحة والعافية .